
XXY (Klinefelter) Syndrome
Overview
Historically, XXY syndrome was called Klinefelter syndrome, which involves a constellation of physical and behavioral characteristics that was first associated with the chromosome complement of XXY by Dr. Harry Klinefelter in the 1940s. With time, many males were found to have the XXY chromosome pattern but not the physical and behavioral phenotypes – it is estimated that only about 25% of men with XXY are ever diagnosed. [Samango-Sprouse: 2019] In In addition to the most typical XXY karyotype, other chromosome abnormalities may lead to a similar presentation. These include XY/XXY mosaicism, where a percentage of cells are normal (46,XY), and a percentage have an extra X chromosome (47,XXY). Individuals with mosaicism may still be fertile, depending on the percentage and distribution of normal XY cells. Although less common, it is also possible to have more than 1 extra X chromosome (e.g., 48,XXXY), which is typically associated with a more severe phenotype and intellectual disability.
Other Names & Coding
Q98.0, Klinefelter syndrome karyotype 47,XXY
Q98.1, Klinefelter syndrome, male with more than 2 X chromosomes
Q98.4, Klinefelter syndrome, unspecified
ICD-10 for Klinefelter Syndrome (icd10data.com) provides further coding details.
Prevalence
Genetics
Prognosis
Practice Guidelines
Although there are no formal guidelines, the following provides an updated
review of XXY syndrome, including causes, diagnostic strategies, and
management.
Samango-Sprouse CA, Counts DR, Tran SL, Lasutschinkow PC, Porter GF, Gropman AL.
Update On The Clinical Perspectives And Care Of The Child With 47,XXY (Klinefelter Syndrome).
Appl Clin Genet.
2019;12:191-202.
PubMed abstract / Full Text
Roles of the Medical Home
Clinical Assessment
Overview
XXY syndrome should be considered in infants and children with developmental delay or in adolescent males who are tall, overweight with fat distributed in a pear-shaped pattern, and have decreased secondary sexual characteristics in puberty, gynecomastia, and learning problems.
Screening
Of Family Members
Presentations
- Infants may be described as "easy" babies. They may present with hypotonia and show developmental delays in some or all areas (particularly language).
- Toddlers may have continued language delays, or they may be diagnosed with sensory integration disorder, auditory processing problems, and autism spectrum disorder.
- Young boys and teens with XXY syndrome may be taller and less muscular than their male peers.
- Adolescents may experience normal or delayed puberty but have decreased facial and body hair; smaller, firmer testicles; decreased muscle development; and gynecomastia. Depression and anxiety may occur at this age; aggressive or oppositional behaviors may also be seen. If hypogonadism is left untreated, affected individuals develop a eunuchoid body habitus, with small testes, gynecomastia, and increased levels of gonadotropins.
- Older teens and adults may not look different from other males their age, except possibly taller. Low levels of testosterone can cause infertility despite normal sexual function and a high-pitched voice. Osteoporosis is often found in older teens and adults with XXY syndrome.
- Hand tremors
- Decreased activity and endurance level
- Sleep abnormalities
- Difficulties in social situations (shy, immature, passive) with low self-esteem
- Learning problems
- Expressive greater than receptive language problems
- Decreased auditory recall
- Easily frustrated
- Higher risk of autoimmune diseases in adults
- Joint laxity
Diagnostic Criteria
Differential Diagnosis
Fragile X syndrome: Those with fragile X may have some of the same physical characteristics that those with XXY have, including intellectual disability, autistic features, hypotonia, and tall stature. The 2 conditions may seem more similar in younger children; in general, they can be distinguished much more easily in older children and adolescents. They can be differentiated definitively by genetic testing, which will demonstrate sex chromosome aneuploidy in individuals with XXY. The Portal’s module on Fragile X Syndrome provides further diagnostic and management information.
Marfan syndrome: Individuals with Marfan syndrome, a connective tissue disorder, are tall of stature and may demonstrate hypotonia. Karyotyping will identify males with the XXY syndrome; Marfan syndrome is a clinical or molecular diagnosis based on family history and affected body systems. It is sometimes confirmed with genetic testing for mutations in the FBN1 gene. See Marfan Syndrome (GeneReviews) for more information.
Other overlapping conditions: Especially in younger children, XXY syndrome has a nonspecific presentation that overlaps with many other genetic conditions, including microdeletion and microduplication syndromes, metabolic disorders such as homocystinuria, and overgrowth disorders such as Sotos syndrome. If routine chromosome studies are normal in a child with features of XXY syndrome, refer for genetic evaluation.
History & Examination
Developmental & Educational Progress
Maturationalprogress
Social & Family Functioning
Physical Exam
General
Growth Parameters
Ht | Wt | BMI Boys are usually tall and may have abnormal body proportions with long legs and a shorter trunk. They can have decreased muscle mass and are often overweight. Weight and BMI should be monitored closely with preventative measures taken to avoid obesity.
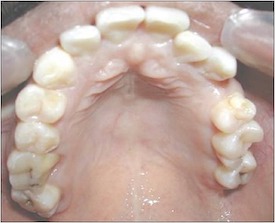
HEENT/Oral
Enamel defects may also be
present. [Ramasamy: 2009]
Genitalia
Neurologic Exam
Neurologic exam should be normal, yet hypotonia and a resting hand tremor are sometimes present. Although strength may be decreased in males with XXY syndrome, this is difficult to demonstrate on exam. [Ross: 2008]
Testing
Laboratory Testing
Genetic Testing
Other Testing
Specialty Collaborations & Other Services
Medical Genetics (see NW providers [1])
Developmental - Behavioral Pediatrics (see NW providers [1])
Pediatric Endocrinology (see NW providers [1])
Treatment & Management
Pearls & Alerts for Treatment & Management
Fertility treatmentsSperm extractions, in conjunction with testosterone therapy, have been used with success to allow fertility in men with XXY syndrome. The treatments are expensive and additional measures, such as intracytoplasmic sperm injection, may also be needed.
Systems
Endocrine/Metabolism
Treatment is usually initiated at 11 or 12 years of age with testosterone enanthate (Delatestryl) or cypionate (Depo-Testosterone) at 25-50 mg IM q 2 weeks–1 month. It is then increased gradually depending on the degree of virilization, growth, and serum gonadotropin levels, which should decrease with therapy to the high normal range. (Final adult dose is approximately 200 mg IM q 2–3 weeks.)
Once treatment starts, monitor for possible side effects including worsening acne, too rapid sexual development, prostate enlargement, and behavior problems–especially aggression. Transdermal methods of testosterone administration may also be used in older individuals with XXY syndrome.
Note that testosterone therapy does not change the rate of infertility. The option of semen collection and sperm-banking (cryopreservation) in XXY individuals during puberty may be discussed during visits to the geneticist or endocrinologist. Sperm is retrievable from about 50% of adolescents/young adults [Nahata: 2016] and from smaller proportions as individuals age.
Specialty Collaborations & Other Services
Pediatric Endocrinology (see NW providers [1])
Development (general)
Specialty Collaborations & Other Services
Developmental - Behavioral Pediatrics (see NW providers [1])
Physical Therapy (see NW providers [0])
Occupational Therapy (see NW providers [1])
Speech - Language Pathologists (see NW providers [4])
Learning/Education/Schools
Specialty Collaborations & Other Services
Mental Health Evaluation/Assessment (see NW providers [0])
Maturation/Sexual/Reproductive
Specialty Collaborations & Other Services
Pediatric Urology (see NW providers [0])
Mental Health/Behavior
The personality and behavior of boys with XXY syndrome have been described as shy, reserved, sensitive, and passive in childhood, which, together with unattended learning difficulties, may lead to secondary adaptive and behavioral problems in adolescence. [Sørensen: 1992] [Ross: 2008] Testosterone treatment from puberty may assist in psychosocial adaption during adolescence. [Simm: 2006] [Maggi: 2007] Social skills training, special education services, or provisions in an individualized educational plan for learning problems may also help improve self-esteem. [Samango-Sprouse: 2002] Sometimes, having a diagnosis can help qualify children for services. More details about the personal impact of XXY syndrome and the influence of age at diagnosis, clinical, social, and demographic factors on adult quality of life outcomes can be found in [Herlihy: 2011].
Specialty Collaborations & Other Services
Developmental - Behavioral Pediatrics (see NW providers [1])
General Counseling Services (see NW providers [1])
Transitions
Ask the Specialist
Should all boys with gynecomastia be tested for XXY syndrome?
Gynecomastia is common among adolescent boys. If puberty is otherwise proceeding normally, including normal testicular growth, XXY syndrome is unlikely.
Can I send testing for XXY syndrome, or does it need to be sent by a specialist?
A routine chromosome study can usually be sent from a primary care provider’s office. Most insurance companies require prior authorization for genetic testing.
Is a karyotype (G-banded chromosome study) or a microarray the best test for XXY syndrome?
It depends on how the patient presents. Either test will detect XXY syndrome. If you have a high concern for 47,XXY and don’t suspect another condition (as in a teenager with gynecomastia and small testes for Tanner stage), a karyotype will probably be sufficient. The microarray, though more expensive, is a more sensitive test for other chromosome abnormalities. It can be considered as a first step in boys with developmental delay or as a second step for children whose chromosome study is normal, but XXY is still suspected. Prior authorization may be needed for these tests.
Resources for Clinicians
On the Web
Klinefelter Syndrome (NORD)
Information for families that includes synonyms, signs & symptoms, causes, affected populations, related disorders, diagnosis,
therapies (both standard and investigational), and support organizations; National Organization of Rare Disorders.
Helpful Articles
PubMed search for XXY (Klinefelter) syndrome in children, last 3 years.
Samango-Sprouse CA, Counts DR, Tran SL, Lasutschinkow PC, Porter GF, Gropman AL.
Update On The Clinical Perspectives And Care Of The Child With 47,XXY (Klinefelter Syndrome).
Appl Clin Genet.
2019;12:191-202.
PubMed abstract / Full Text
Updated review of Klinefelter syndrome, including causes, diagnostic strategies and management
Shiraishi K, Matsuyama H.
Klinefelter syndrome: From pediatrics to geriatrics.
Reprod Med Biol.
2019;18(2):140-150.
PubMed abstract / Full Text
Recommendations on how to transition patients with Klinefelter syndrome from pediatrics to adult care.
Bearelly P, Oates R.
Recent advances in managing and understanding Klinefelter syndrome.
F1000Res.
2019;8.
PubMed abstract / Full Text
Review on the current standard of care in terms of management for patients with Klinefelter syndrome.
Resources for Patients & Families
Information on the Web
Klinefelter Syndrome (MedlinePlus)
Information for families that includes description, frequency, causes, inheritance, other names, and additional resources;
from the National Library of Medicine.
Klinefelter Syndrome (MedlinePlus)
Information for families that includes description, frequency, causes, inheritance, other names, and additional resources;
from the National Library of Medicine.
Klinefelter Syndrome (MAYO)
Includes information about symptoms, inheritance, diagnosis, finding a specialist, related diseases, and support organizations
MAYO Clinic
National & Local Support
American Association for Klinefelter Syndrome Information and Support (AAKSIS)
Information, education, and support via toll-free telephone line, e-mail, network of support groups, and an annual educational
conference.
Association for X and Y Variations (AXYS)
An organization founded by a mother of a child with Klinefelter syndrome that provides resources, research, and support for
those affected by X and Y chromosome variations.
Studies/Registries
Clinical trials in XXY (Klinefelter) syndrome (clinicaltrials.gov)
Studies looking at better understanding, diagnosing, and treating this condition; from the National Library of Medicine.
Services for Patients & Families Nationwide (NW)
| Service Categories | # of providers* in: | NW | Partner states (4) (show) | | NM | NV | RI | UT | |
|---|---|---|---|---|---|---|---|---|---|
| Developmental - Behavioral Pediatrics | 1 | 2 | 3 | 12 | 9 | ||||
| Family Counseling | 1 | 23 | 44 | 69 | |||||
| General Counseling Services | 1 | 10 | 213 | 30 | 298 | ||||
| Medical Genetics | 1 | 2 | 5 | 4 | 7 | ||||
| Mental Health Evaluation/Assessment | 8 | 9 | 24 | 131 | |||||
| Occupational Therapy | 1 | 17 | 22 | 22 | 37 | ||||
| Pediatric Endocrinology | 1 | 4 | 6 | 12 | 7 | ||||
| Pediatric Urology | 13 | 1 | 3 | ||||||
| Physical Therapy | 12 | 9 | 7 | 40 | |||||
| Speech - Language Pathologists | 4 | 23 | 11 | 34 | 65 | ||||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Bibliography
Bearelly P, Oates R.
Recent advances in managing and understanding Klinefelter syndrome.
F1000Res.
2019;8.
PubMed abstract / Full Text
Review on the current standard of care in terms of management for patients with Klinefelter syndrome.
Herlihy AS, McLachlan RI, Gillam L, Cock ML, Collins V, Halliday JL.
The psychosocial impact of Klinefelter syndrome and factors influencing quality of life.
Genet Med.
2011;13(7):632-42.
PubMed abstract
Investigates the personal impact of Klinefelter syndrome and the influence of age at diagnosis, clinical, social, and demographic
factors on adult quality of life outcomes.
Jayashankara C, Shivanna AK, Sridhara K, Kumar PS.
Taurodontism: A dental rarity.
J Oral Maxillofac Pathol.
2013;17(3):478.
PubMed abstract / Full Text
Maggi M, Schulman C, Quinton R, Langham S, Uhl-Hochgraeber K.
The burden of testosterone deficiency syndrome in adult men: economic and quality-of-life impact.
J Sex Med.
2007;4(4 Pt 1):1056-69.
PubMed abstract
Morris JK, Alberman E, Scott C, Jacobs P.
Is the prevalence of Klinefelter syndrome increasing?.
Eur J Hum Genet.
2008;16(2):163-70.
PubMed abstract
Nahata L, Yu RN, Paltiel HJ, Chow JS, Logvinenko T, Rosoklija I, Cohen LE.
Sperm Retrieval in Adolescents and Young Adults with Klinefelter Syndrome: A Prospective, Pilot Study.
J Pediatr.
2016;170:260-5.e1-2.
PubMed abstract
Ramasamy R, Ricci JA, Palermo GD, Gosden LV, Rosenwaks Z, Schlegel PN.
Successful fertility treatment for Klinefelter's syndrome.
J Urol.
2009;182(3):1108-13.
PubMed abstract
Ross JL, Roeltgen DP, Stefanatos G, Benecke R, Zeger MP, Kushner H, Ramos P, Elder FF, Zinn AR.
Cognitive and motor development during childhood in boys with Klinefelter syndrome.
Am J Med Genet A.
2008;146A(6):708-19.
PubMed abstract
Samango-Sprouse, C.
XXY: The Hidden Disability and a Prototype for an Infantile Presentation of Developmental Dyspraxia (IDD.
Infants Young Child.
2002(15):11-18.
PubMed abstract
Samango-Sprouse CA, Counts DR, Tran SL, Lasutschinkow PC, Porter GF, Gropman AL.
Update On The Clinical Perspectives And Care Of The Child With 47,XXY (Klinefelter Syndrome).
Appl Clin Genet.
2019;12:191-202.
PubMed abstract / Full Text
Updated review of Klinefelter syndrome, including causes, diagnostic strategies and management
Shiraishi K, Matsuyama H.
Klinefelter syndrome: From pediatrics to geriatrics.
Reprod Med Biol.
2019;18(2):140-150.
PubMed abstract / Full Text
Recommendations on how to transition patients with Klinefelter syndrome from pediatrics to adult care.
Simm PJ, Zacharin MR.
The psychosocial impact of Klinefelter syndrome--a 10 year review.
J Pediatr Endocrinol Metab.
2006;19(4):499-505.
PubMed abstract
Sørensen K.
Physical and mental development of adolescent males with Klinefelter syndrome.
Horm Res.
1992;37 Suppl 3:55-61.
PubMed abstract
Visootsak J, Graham JM Jr.
Klinefelter syndrome and other sex chromosomal aneuploidies.
Orphanet J Rare Dis.
2006;1:42.
PubMed abstract / Full Text
Zeger MP, Zinn AR, Lahlou N, Ramos P, Kowal K, Samango-Sprouse C, Ross JL.
Effect of ascertainment and genetic features on the phenotype of Klinefelter syndrome.
J Pediatr.
2008;152(5):716-22.
PubMed abstract