
Tyrosinemia Type 1
Guidance for primary care clinicians receiving a positive newborn screen result
Screening
Tested By
Description
Tyrosine Catabolic Pathway
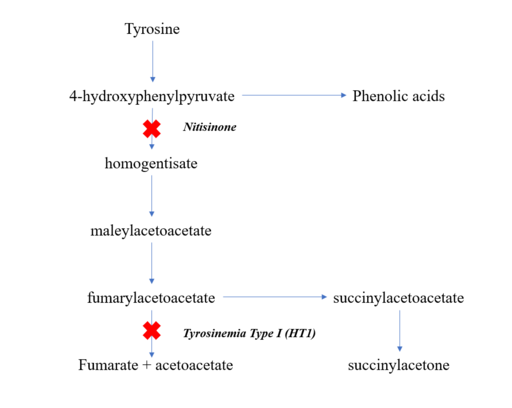
With time, elevated succinylacetone and related toxic compounds produced in the liver and kidneys will cause liver and renal dysfunction, failure to thrive, rickets due to severe renal tubular dysfunction, and high risk for developing hepatocellular carcinoma. This most severe form is usually asymptomatic in the neonate.
Clinical Characteristics
Without treatment, chronic problems ensue, including liver disease leading to cirrhosis and hepatocellular carcinoma, renal tubular disease (Fanconi syndrome), rickets, failure to thrive, and coagulation disorders. Repeated neurologic crises may involve mental status change, abdominal pain, peripheral neuropathy, and/or respiratory failure. These are due to the accumulation of delta-aminolevulinic acid, whose metabolism is inhibited by succinylacetone. Death usually occurs by 10 years of age.
- Severe liver involvement in the young infant with:
- Jaundice
- Ascites
- Coagulopathy
- GI bleeding
- Or development later in the first year of life:
- Liver dysfunction
- Renal involvement
- Growth failure
- Rickets
- "Boiled cabbage" or "rotten mushroom" odor to the body and urine
Incidence
Primary Care Management
Next Steps After a Positive Screen
- Contact the family and evaluate the infant for evidence of liver disease, jaundice, diarrhea, or vomiting (although most neonates will be asymptomatic).
- Provide emergency treatment/referral for jaundice, bloody stools, or
seizures. See the ACT Sheet for Tyrosinemia (ACMG) (
 348 KB).
348 KB).
Confirming the Diagnosis
- To confirm the diagnosis, work with Newborn Screening Services (see NW providers [1]).
- Quantitative plasma amino acid analysis, urine organic acid analysis. Quantitative succinyl acetone may be advised to differentiate from the other forms of tyrosinemia if not included in the newborn screening process. Molecular genetic testing may need to be obtained to confirm the diagnosis.
If the Diagnosis is Confirmed
- For evaluation and ongoing collaborative management, consult Medical Genetics (see NW providers [1]).
- Educate the family regarding signs, symptoms, and the need for urgent care when the infant becomes ill (see Tyrosinemia Type 1 - Information for Parents (STAR-G) ).
- Support initiation and maintenance of dietary restriction of phenylalanine and tyrosine.
- Vitamin D supplements may be indicated for affected children. See Calcium and Vitamin D.
- Regular blood and urine tests to monitor amino acid, succinylacetone, and alpha-fetoprotein levels; kidney and liver function; and diet may be indicated.
- Abdominal ultrasounds are performed on a regular basis, with an MRI of the liver performed when there are suspicious lesions.
- For those identified after irreversible sequelae, assist in management of liver and kidney disease and support with developmental and educational interventions. Refer infants at risk for developmental delays for assessment and support to Early Intervention for Children with Disabilities/Delays (see NW providers [3]).
- Initiate consultation and ongoing collaboration with gastroenterology and nephrology if the child is affected.
- Nutrition, Metabolic (see NW providers [11]) may work with the family to devise an optimal approach to dietary management.
- Refer the family to Genetic Testing and Counseling (see NW providers [6]).
Resources
Information & Support
Related Portal Content
Tyrosinemia Type 1
Assessment and management information for the primary care
clinician caring for the child with Tyrosinemia Type 1.
After a Diagnosis or Problem is Identified
Families can face a big change when their baby tests positive for
a newborn condition. Find information about A New Diagnosis - You Are Not Alone;
Caring for Children with Special Health Care Needs; Assistance in Choosing
Providers; Partnering with Healthcare Providers; Top Ten Things to Do After a
Diagnosis.
For Professionals
Tyrosinemia Type 1 (GeneReviews)
Clinical characteristics, diagnosis/testing, management, genetic counseling, and molecular pathogenesis; from the University
of Washington and the National Library of Medicine.
Tyrosinemia Type 1 (OMIM)
Information about clinical features, diagnosis, management, and molecular and population genetics; Online Mendelian Inheritance
in Man.
Newborn Screening (HRSA)
Detailed information about the newborn screening process by state and condition; Health Resources & Services Administration.
Tyrosinemia (Canadian Liver Foundation)
Cause, symptoms, diagnosis, and treatment information.
For Parents and Patients
Tyrosinemia Type 1 - Information for Parents (STAR-G)
A fact sheet, written by a genetic counselor and reviewed by metabolic and genetic specialists, for families who have received
an initial diagnosis of this newborn disorder; Screening, Technology and Research in Genetics.
Tyrosinemia Type 1 (GARD)
Includes information about symptoms, inheritance, diagnosis, finding a specialist, related diseases, and support organizations;
Genetic and Rare Diseases Information Center of the National Center for Advancing Translational Sciences.
Practice Guidelines
Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G, Waisbren SE, Gucsavas-Calikoglu M, Wasserstein
MP, Coakley K, Scott CR.
Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations.
Genet Med.
2017;19(12).
PubMed abstract / Full Text
Tools
ACT Sheet for Tyrosinemia (ACMG) ( 348 KB)
Provides recommendations for clinical and laboratory follow-up of the newborn with out-of-range screening results, along with
national for clinicians and families; American College of Medical Genetics.
Confirmatory Algorithm for Tyrosinemia (ACMG) ( 146 KB)
An algorithm of the basic steps involved in determining the final diagnosis of an infant with a positive newborn screen; American
College of Medical Genetics.
Services for Patients & Families Nationwide (NW)
| Service Categories | # of providers* in: | NW | Partner states (4) (show) | | NM | NV | RI | UT | |
|---|---|---|---|---|---|---|---|---|---|
| Early Intervention for Children with Disabilities/Delays | 3 | 34 | 30 | 13 | 51 | ||||
| Genetic Testing and Counseling | 6 | 6 | 12 | 8 | 11 | ||||
| Medical Genetics | 1 | 2 | 5 | 4 | 7 | ||||
| Newborn Screening Services | 1 | 3 | 2 | 2 | 3 | ||||
| Nutrition, Metabolic | 11 | 11 | 13 | 13 | 11 | ||||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Studies
Tyrosinemia Type 1 (clinicaltrials.gov)
Studies looking at better understanding, diagnosing, and treating this condition; from the National Library of Medicine.
Helpful Articles
PubMed search for tyrosinemia type 1 and neonatal screening, last 10 years
de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, de Baulny HO, Pintos-Morell G, Spiekerkötter U.
Recommendations for the management of tyrosinaemia type 1.
Orphanet J Rare Dis.
2013;8:8.
PubMed abstract / Full Text
Developed by a European collaboration; recommendations may not apply to the United States, particularly as they relate to
early diagnosis, since tyrosinemia is now routinely screened for in the US.
McKiernan PJ, Preece MA, Chakrapani A.
Outcome of children with hereditary tyrosinaemia following newborn screening.
Arch Dis Child.
2015;100(8):738-41.
PubMed abstract
van Ginkel WG, Rodenburg IL, Harding CO, Hollak CEM, Heiner-Fokkema MR, van Spronsen FJ.
Long-Term Outcomes and Practical Considerations in the Pharmacological Management of Tyrosinemia Type 1.
Paediatr Drugs.
2019;21(6):413-426.
PubMed abstract / Full Text
Pass K, Morrissey M.
Clinical Chemistry: Enhancing Newborn Screening for Tyrosinemia Type I.
Volume 54, Issue 4 ed. pp. 627–629: Oxford Academic;
2008.
https://academic.oup.com/clinchem/article/54/4/627/5628481
Page Bibliography
Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G, Waisbren SE, Gucsavas-Calikoglu M, Wasserstein
MP, Coakley K, Scott CR.
Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations.
Genet Med.
2017;19(12).
PubMed abstract / Full Text
de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, de Baulny HO, Pintos-Morell G, Spiekerkötter U.
Recommendations for the management of tyrosinaemia type 1.
Orphanet J Rare Dis.
2013;8:8.
PubMed abstract / Full Text
Developed by a European collaboration; recommendations may not apply to the United States, particularly as they relate to
early diagnosis, since tyrosinemia is now routinely screened for in the US.
Fuenzalida K, Leal-Witt MJ, Guerrero P, Hamilton V, Salazar MF, Peñaloza F, Arias C, Cornejo V.
NTBC Treatment Monitoring in Chilean Patients with Tyrosinemia Type 1 and Its Association with Biochemical Parameters and
Liver Biomarkers.
J Clin Med.
2021;10(24).
PubMed abstract / Full Text
McKiernan PJ, Preece MA, Chakrapani A.
Outcome of children with hereditary tyrosinaemia following newborn screening.
Arch Dis Child.
2015;100(8):738-41.
PubMed abstract
Pass K, Morrissey M.
Clinical Chemistry: Enhancing Newborn Screening for Tyrosinemia Type I.
Volume 54, Issue 4 ed. pp. 627–629: Oxford Academic;
2008.
https://academic.oup.com/clinchem/article/54/4/627/5628481
Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF.
Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome,
and implications.
Pediatrics.
2003;111(6 Pt 1):1399-406.
PubMed abstract
Therrell BL Jr, Lloyd-Puryear MA, Camp KM, Mann MY.
Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions
for research agenda planning.
Mol Genet Metab.
2014;113(1-2):14-26.
PubMed abstract / Full Text
van Ginkel WG, Rodenburg IL, Harding CO, Hollak CEM, Heiner-Fokkema MR, van Spronsen FJ.
Long-Term Outcomes and Practical Considerations in the Pharmacological Management of Tyrosinemia Type 1.
Paediatr Drugs.
2019;21(6):413-426.
PubMed abstract / Full Text